Website Introduction and Brief Summary
In a time of universal deceit - telling the
truth is a revolutionary act. (George Orwell)
You must be the change you wish to see in the world. (Gandhi)
All that is necessary for evil to succeed is for good men to do nothing.
(Edmund Burke)
Hell is Truth Seen Too Late. (Thomas Hobbes)
 Hello
Everyone - We have set this page up as a 'blog' of our Introductions. So
though the page may seem a little long, it just contains five introductions
to this website that date back to 2005. They are perhaps all a bit abrupt
(but we realise how busy people are, a curse of the modern world!). We hope
that together they provide a good overview of what this site is about, why
we think it is important.
Hello
Everyone - We have set this page up as a 'blog' of our Introductions. So
though the page may seem a little long, it just contains five introductions
to this website that date back to 2005. They are perhaps all a bit abrupt
(but we realise how busy people are, a curse of the modern world!). We hope
that together they provide a good overview of what this site is about, why
we think it is important.
At the bottom of the page there is a nice collection of quotes that we hope
will motivate you to get involved and help save our planet as a way of protecting
your life and the lives of your children.
With kindness, beauty and truth,
Geoff Haselhurst
Site Introduction (2012): Despite several thousand years of failure to correctly understand physical reality (hence the current postmodern view that this is impossible) there is an obvious solution.
Simply unite Science (Occam's Razor / Simplicity) with Metaphysics (Dynamic Unity of Reality) and describe reality from only one substance existing, as Leibniz wrote;
'Reality cannot be found except in One single source, because of the interconnection of all things with one another'.
Given we all experience many minds and many material things, but always in one common Space, we are thus required to describe physical reality in terms of Space. We then find there is only one solution, a Wave Structure of Matter (WSM) where the electron is a spherical standing wave. See Wave Diagrams.
In hindsight the error was obvious, to try and describe an interconnected reality with discrete 'particles', which then required forces / fields to connect them in space and time. This was always just a mathematical solution which never explained how matter was connected across the universe.
I realise that there are a lot of 'crackpot' theories about truth and reality on the internet, but it is easy to show that the Wave Structure of Matter is the correct solution as it deduces the laws of Nature (the fundamentals of Physics & Philosophy) perfectly (there are no opinions). While the Wave Structure of Matter is obvious once known, to begin it will seem strange simply because it takes time for our minds to adjust to new knowledge.
For those who are religious / spiritual, I think Albert Einstein expresses the enlightened view of God. He writes 'I believe in Spinoza's God who reveals himself in the orderly harmony of what exists, not in a God who concerns himself with the fates and actions of human beings.' This harmony arises from a Wave Structure of Matter in Space (we are all interconnected in this space that we all commonly experience). This unity of reality (God, Brahman, Tao, Spirit, Energy, Light, Vibration) is central to all major world religions, thus their common moral foundation of 'Do unto others as to thyself' as the other is part of the self.
Please help our world (human society / life on earth) by sharing this knowledge.
Clearly our world is in great trouble due to human overpopulation and the resultant destruction of Nature, climate change and the pollution of air, land and water. The best solution to these problems is to found our societies on truth and reality rather than past myths and customs (which invariably cause harm).
Given the Censorship in Physics / Philosophy of Science Journals (founded on the standard model / particle physics) the internet is clearly the best way to get new knowledge visible to the world.
A world now in great need of wisdom from truth and reality.
Sincerely,
Geoff Haselhurst - - Full Introduction - Email - Nice Letters - Share this Knowledge
In a time of universal deceit - telling the truth is a revolutionary act. (George Orwell)
You must be the change you wish to see in the world. (Mohandas Gandhi)
All that is necessary for evil to succeed is for good men to do nothing. (Edmund Burke)
Hell is Truth Seen Too Late. (Thomas Hobbes)
Introduction (Jan. 2009)
Hello and welcome! Please excuse the abruptness of this introduction - it is written with Albert Einstein's ideals of kindness, beauty and truth in mind.
I am convinced that we have finally worked out what physical reality is. i.e. We can correctly imagine how matter exists and moves about in Space (including you and I and all other life on earth, all those stars across the universe). This website explains why I am certain this is true - why I think this knowledge is vital for the future survival of humanity.
I appreciate that after several thousand years of failure to understand
truth and reality it is now generally accepted by our skeptical
postmodern world that true knowledge of reality is impossible. However
the solution was simple and obvious once known. We just had to take Science
(Occam's Razor / Simplicity) and Metaphysics
(Dynamic Unity of Reality) seriously and thus describe reality from
only one substance existing, as Leibniz wrote;
Reality cannot be found except in One single source,
because of the interconnection of all things with one another.
When we deduce
the most Simple Science Theory of Reality we find that there is only
one possible solution:
Space must be the substance which exists and matter is formed from
waves in Space.
In hindsight the error was obvious - to try and describe
an interconnected
reality from the foundation of many discrete and separate things, matter
'particles' moving around in space and time, which then required the invention
of continuous forces / fields to connect the discrete 'particles'. This
was always just a mathematical
approach which never explained how matter was physically interconnected
in Space.
Conversely, the wave structure of matter (by describing reality in terms
Space, the one thing all matter exists in) enables us to clearly picture
how matter is interconnected. The spherical IN and OUT waves interact with
all other wave-center 'particles' in the universe. Thus matter is in continual
two way communication with all the matter around it (matter is active and
dynamic) which explains both how it can move, and the true cause of time.
 An
electron is simply a spherical standing wave in space. The positron (antimatter)
is just the opposite phase wave.
An
electron is simply a spherical standing wave in space. The positron (antimatter)
is just the opposite phase wave.
We can then show that this is the correct solution, as the Wave Structure
of Matter (WSM) in Space deduces the fundamentals of Physics
& Philosophy.
i.e. Once you know reality it is obvious that it is correct as it explains
so many things so perfectly. And the solutions are deduced, there are no
opinions.
I realise that there are a lot of 'crackpot' theories about truth and reality
on the internet, but you can work this out for yourselves and you will see
that it is correct. It is the most simple solution, it works perfectly and
it is obvious
once known (though it does take a while for our minds to adjust to new
knowledge).
Science does work, we just needed the correct (most simple) foundation of
space and its wave motions (rather than the motion of matter particles and
their connecting forces / fields in space and time). Physical reality (what
we really are as humans) is obviously important, it is the source of all
truth and wisdom. But this knowledge only helps society if the majority
are aware of it.
Please help
our world (human society / life on earth) by sharing these pages. We
are creating hell on earth by destroying Nature (a part of our true selves).
-----------------------------------------------------------------
Why Space is the most simple foundation for describing
the world we all experience.
Imagine driving your car on a busy road. There are obviously many other
people driving their cars, thus many minds and material things, but they
all interact in the same space. This is true when crowds of people collectively
watch some event (e.g. fireworks, a sporting event, a lunar eclipse, the
distant stars), we always experience many minds and material things, but
always in one common Space.
Thus Space is the most simple foundation for Science, as it is always perceived
as the one thing that we all commonly experience.
So now we must consider the Properties of Space, as Aristotle
and Leibniz
explained. Given the particle / wave duality of light and matter, and that
we cannot add 'particles' to Space then there is only one solution: Space
has the properties of a wave medium and matter is formed from wave motions
of Space. Further, we know that matter interacts spherically from Einstein's
relativity, thus the most
simple solution is that matter is formed from spherical standing waves
in Space.
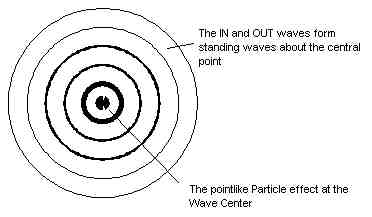
So how do Waves explain the 'particle' properties of light and matter? Simple. The particle effect of matter is formed at the wave-center of the spherical standing wave (see diagram) and the particle effect of light is due to resonance and discrete frequency interactions where energy E=hf (Planck's constant by frequency). Basically quantum physics is founded on wave equations, and general relativity on the mathematics for the curvature of a sphere (where matter curves space-time). So you can see that the spherical wave structure of matter unites these two central concepts of physics (Einstein's spherical geometry and quantum waves) perfectly.
What about time?
Well it is really a human construction, there is just the present now (space),
but it is in a state of change (space is vibrating if you prefer). The future
is just the In waves flowing through space that will form your future wave
center 'particles', the past is your out waves.
For those who are religious
/ spiritual, you can consider space as God (Brahman, Tao, Spirit, Energy,
Light). What is certain is that discrete and separate 'particles'
do not exist - we are all connected to this space that we all commonly experience.
And this underlying unity of reality (God) is central to all major world
religions, thus their common moral foundation of 'Do unto others as to thyself'
(as the other is part of the self / we are all one with god).
While I am certain that matter is formed from waves in space, there is still
much to explain about our minds, our human emotions and moral / spiritual
sense. This is no doubt the future of theology and our understanding of
God, to explore the properties of this Space we all find ourselves existing
in.
Understanding this underlying unity of reality is now critically
important to humanity. Our world is in great trouble, heading rapidly
towards self destruction due to human overpopulation and the resultant destruction
of Nature,
climate
change and the pollution of air, land and water, contaminating everything
we consume. Clearly the best solution to these problems is to found
our societies on truth and reality rather than past myths and customs
which invariably cause conflict and harm.
Support
an open honest (sensible logical) science discussion of Truth & Reality
(anchor link to help section lower on this page).
I am not naive to the difficulties that humanity faces. But history shows
that truth eventually triumphs, that there are enough sensible logical people
in the world who appreciate the importance of truth.
Given the
Censorship in Physics / Philosophy of Science Journals (founded on the
standard model / particle physics) the internet is clearly the best way
to get new knowledge visible to the world.
A world now in great need of wisdom from truth and reality.
Sincerely,
Geoff
Haselhurst - - Email
- Nice
letters we receive
A Short Summary
For the past 350 years (since Newton) physics has been misled by focusing
on the 'particle' structure of matter in 'space-time',
rather than the more simple Wave Structure of Matter in Space.
i.e. The mistake of studying light and matter 'particles' rather than the
Space they exist in.
 Quantum
Physics: The Wave Structure of Matter (WSM) provides a simple sensible
solution to the particle-wave duality of light and matter.
Quantum
Physics: The Wave Structure of Matter (WSM) provides a simple sensible
solution to the particle-wave duality of light and matter.
As Schrodinger wrote, 'What we observe as material bodies
and forces are nothing but shapes and variations in the structure of space.
Particles are just schaumkommen (appearances).'
The 'particle' effect of matter is caused by the Wave-Center of the
Spherical Standing Waves in Space (see Wave
Diagrams).
The discrete 'photon particle' properties of light are caused by standing
wave interactions (resonant coupling) which only occur at discrete frequencies
(thus discrete energy states, where E=hf). This is why Schrodinger's Wave
Equations are used to determine the allowed energy / wave states of electrons
in matter - because matter is made of waves (obviously!).
 Albert
Einstein's Theory of Relativity also correctly realized that matter
was large, a structure of space / the universe (there were no discrete little
'particles' which require 'continuous fields' in 'space-time' to connect
them).
Albert
Einstein's Theory of Relativity also correctly realized that matter
was large, a structure of space / the universe (there were no discrete little
'particles' which require 'continuous fields' in 'space-time' to connect
them).
He writes, 'When forced to summarize the general theory
of relativity in one sentence: Time and space and gravitation have no separate
existence from matter. ... since physical reality is represented by a continuous
field, the concept of particles or material points cannot play a fundamental
part, ... and can only appear as a limited region in space in which the
field strength or the energy density are particularly high.' (Albert Einstein)
Einstein just made the mistake of working from a continuous field
theory of matter-energy in space-time, rather than the discrete standing
Wave Structure of Matter in Space (which is why his continuous
field theory failed, as it could never explain the discrete 'particle' properties
of light and matter caused by discrete standing wave interactions / resonant
coupling).
 Cosmology.
If we start with this most simple foundation that only one thing exists,
space, then it is necessarily infinite and eternal. From this foundation
of our finite observable universe being part of infinite space we can then
show that you get a redshift
with distance due to decreasing overlap of Hubble spheres (finite observable
universe). Basically every wave center 'particle' is at the center of its
finite spherical universe within infinite space - and as matter moves further
apart there is less overlap of Hubble spheres, thus less wave interactions
which leads to a redshift with distance.
Cosmology.
If we start with this most simple foundation that only one thing exists,
space, then it is necessarily infinite and eternal. From this foundation
of our finite observable universe being part of infinite space we can then
show that you get a redshift
with distance due to decreasing overlap of Hubble spheres (finite observable
universe). Basically every wave center 'particle' is at the center of its
finite spherical universe within infinite space - and as matter moves further
apart there is less overlap of Hubble spheres, thus less wave interactions
which leads to a redshift with distance.
Further, we can then deduce Mach's
principle and unite
finite matter with infinite space due to Huygens' principle / sharing of
waves (this is very important - one of the great deductions from Milo's
Wolff's pioneering work on the Wave Structure of Matter).
I have spent twelve years thinking on the Wave Structure of Matter (WSM),
while reading the history of physics, philosophy and metaphysics. I have
met (over the internet) an emerging group of scientists and philosophers
(most importantly Dr Milo Wolff) who have also independently discovered
the Wave Structure of Matter, share this same certainty that it is correct
due to its simplicity and power to explain many things, that this knowledge
is important to Humanity.
It is now clear that all matter interactions are really wave interactions
in Space. We humans, and everything we experience around us (the earth and
its life, the sun and stars) all exist as very complexly evolved wave centers
of (spherically) vibrating space - all intimately interconnected to one
another by our spherical in and out waves in space (which is why you can
see stars across the universe, as they are a part of you, and you are a
part of them - a nice thought really).
Separate and discrete 'bodies' made of 'particles' are an illusion of the
mind and how it represents our senses - the fact that we have senses tells
us we are connected to things around us in Space. It is just that most wave
interactions occur at the high wave amplitude wave center (of the spherical
standing waves), so we have evolved to see these high amplitude wave center
'particles'.
True Knowledge of Reality will transform Humanity. By understanding how
things are necessarily interconnected (which is central to all science /
knowledge) we can determine which of our ideas correspond to real things
that exist Vs. imaginary things (e.g. dragons, particles). This certainty
of truth will end the current 'postmodern relativistic' (social construct
/ logical positivist) view of no absolute truths that leaves postmodern
philosophy, physics & metaphysics full of doubt, conflict and confusion.
And clearly these errors in thinking and acting ultimately harm us and our
societies.
Science does work (deduced / no opinions). We can clearly imagine Reality
and the physical existence of Space and its spherical standing wave motions
that cause matter and its interactions.
If you give truth and reality a chance they will repay you many times over,
by providing true guidance (wisdom) for how to think and live on our precious
planet - with much greater harmony amongst ourselves and Nature (the Universe)
from which we evolved, and to which we owe our future existence.
Sincerely,
Geoff
Haselhurst - - Email
- Nice
Letters
Introduction / Summary (Feb. 2007): Hi Everyone.
Firstly, our thanks to the people who have taken the time to write us such
Nice
Letters - we really appreciate it! Below is a brief introduction &
summary to this Physics Philosophy Metaphysics Website which we hope everyone
will read.
Over the past ten years we have read many great minds on the subjects of
truth and reality (physics,
philosophy
and metaphysics)
and collected numerous famous
quotes and images
which make up a large part of these pages. However, we wish to emphasise that the purpose of this
website is not just to present existing knowledge, but rather to show that
despite the postmodern
view of 'no absolute truths', there is actually a very simple
sensible language for understanding physical reality and thus knowing
absolute truth.
(NOTE: It has been a lot of work to build this website so that it gets a large number of visitors. We have done this for two reasons; because we need help to develop this knowledge, and help to add links to these pages so they rank well in Google. The downside is that the website is still a rough draft and will not be complete for several years yet - so some patience and understanding is required!)
So let us now briefly explain this most simple science theory of physical reality and why we think it is correct.
In hindsight the mistake is obvious. We start with Space (empirical senses require Space for things to exist in - we all commonly experience existing in Space). The error was to add 'time' and 'particles' to Space which then led to the problems of how these things are interconnected, thus we had to also add 'forces / fields' to connect the 'particles' in 'space-time'. Newton first formalised this particle conception of matter (which dates back to Ancient Greek Philosophy), and it became the foundation for all of Physics. However, Philosophy and Metaphysics have always known that there was an underlying Dynamic Unity of Reality, to explain the necessary interconnection of all things in the Universe.
The solution is to begin and end with Space (not add other things
to it!). Thus we must consider the Properties of Space. And given
the particle
/ wave duality of light and matter, it then follows that if there are
no 'particles' then we should consider if matter is a Wave Structure of
Space - that Space (which we all commonly experience) exists with the Properties
of a Wave Medium.
Thus the Wave Structure of Matter (WSM) assumes that Space is the one thing
that exists, which causes and connects the many things we experience (matter
as spherical standing waves in space). This is the most Simple
Science Theory for describing physical reality, and it clearly works,
as it deduces (and thus unites) fundamentals of Quantum
Physics, Albert
Einstein's Theory of Relativity and Cosmology
(see also the
Problems of the Big Bang Theory). Anyone can determine the truth of
this for themselves - it is deduced, not anyone's opinion.
Why is this important? Well it tells us what we really are as humans, how we exist as matter in Space, how we are subtly interconnected to all other matter in the universe (which is pretty amazing when you really think about it!). And it corrects past errors of knowledge that currently cause harm not just to the Sciences, but to Society itself. i.e. To those with some knowledge of philosophy and metaphysics, it solves Aristotle's problem of Metaphysics (Prime Mover), by describing the one substance that exists (Space) and its properties (Wave Medium), which then solves David Hume's Problem of Causation and Necessary Connection by explaining how matter is necessarily interconnected to other matter in Space by its spherical In and Out Waves. This is why Science works (how logic and senses can exist), because waves behave logically due to the Properties of Space, and we can sense matter in the space around us because our spherical In Waves interact with other matter waves in the Space around us, and ultimately determine the location of our wave center 'particles'.
And when considered, it is strange / negligent to 'write off' science while the most simple and obvious solution has been ignored (this is why philosophy is known as the discovery of the obvious, as we are often blind to the obvious, history clearly confirms this). Now that this knowledge has been realised, it seems to me that scientists would be silly / negligent to ignore it. Yet the frustrating thing is that most will ignore this knowledge. As I see things (human nature) the main problems are;
i) Several thousand years of failure to work out reality (which is simple and obvious once known), thus most people / academics are now so skeptical that they just assume it is impossible for us to correctly imagine reality (we are trapped in the Mind!). Any mention of absolute truth and reality is deemed 'crackpot'. This is made more difficult again as the internet is full of crackpot theories on truth and reality - and most people are not natural philosophers (including most academic philosophers and physicists) thus they do not have the skills / ability to determine quality from crap, truth from fanciful imagination (this sounds harsh, but it is common observation of many philosophers, from Albert Einstein to Plato and Aristotle).
ii) The current censorship in physics publications which makes it very difficult to get new knowledge published in scientific journals (particularly when it contradicts the existing 'particle' paradigm, despite the general acceptance that particle physics does not work and causes numerous paradoxes & problems). See article by Nobel Prize wining Physicist Brian Josephson on Censorship in Physics Preprint Archive (Science Publications / Journals)
iii) Most academics are now very specialised, very busy, and tend to have a vested interest in maintaining the existing knowledge foundations (since they have built a career on them). This makes it difficult for them to consider new knowledge (even if they wanted to).
iv) People tend to have their own conceptions of truth and reality (which they like), and once humans have formed their ideas they rarely change.
v) We have evolved from primitive tribal (herd like) animals. We are emotional / religious creatures seeking common beliefs which bind us together and enhance the survival of our tribe (we are more likely to fight for a common emotional belief, our history of warfare shows this). Thus our minds are not well suited for thinking carefully about truth and reality (we tend to be confused by our limitless imaginations and incomplete senses, often believe things simply because they are part of our ancestry and culture).
vi) Finally, we only sense a fraction of the real world, which is then represented by our mind (e.g. the sky is not blue, though we do see a certain frequency of light waves as the Wave Structure of Matter explains). Likewise, we 'see' things as separate and discrete objects when reason (and physics) tell us that all things in the universe are subtly interconnected (e.g. to explain why the earth orbits the sun, how we can see stars across the universe).
Given these difficulties, it seems that using the Internet is the most efficient way to get this knowledge visible to the world (thus explaining why this website exists - and why we depend upon your help). History shows that despite our human frailties, true knowledge does slowly filter into society, that there are enough sensible logical people with skeptical open minds who recognise truth when they see it.
Philosophy and Society: Wisdom from Truth and Reality
I am sure that most people realise that the world has many serious and disturbing
problems. e.g. Human overpopulation resulting in pollution of air food and
water, the destruction of Nature (with radical climate change a likely result),
and the endless conflicts that arise when our societies are founded on custom
/ myth rather than truth (our current world / society).
It also seems that we individuals have little power to improve things.
This is something that initially depressed me, I live in Nature
(on 600 acres of natural bush) and find Nature both beautiful and amazing.
It is clearly true that we evolved from Nature and depend upon Nature
for survival, yet as a collective we are obviously destroying Nature, thus
our future survival (stop and think about your children for a moment,
their future).
History shows us though, that it is we individuals who do actually change
the world, because it is individuals who discover knowledge, and it is knowledge
that changes the world by changing how we think and live. Where
we have true knowledge we tend to act more wisely (philosophy is
founded on this), when our knowledge is untrue (myth, custom) we are more
foolish, stupid and inclined to conflicting 'cultural / relative truths'
that cause Humanity endless conflicts and brutality.
Introduction
The Metaphysics of Space & the Wave Structure
of Matter
I must admit that I have spent considerable time thinking about writing
an introduction to this website on Truth and Reality. As
a philosopher these concepts are central to all the work I do. Unfortunately,
I happen to live at a time where 'reality' is more closely associated with
'reality TV', and it seems everyone is entitled to their own truths, no
matter how fanciful they may be (the joy of 'relative truth'). It is almost
deemed insulting to some, to suggest that there is a physical reality, and
as a consequence, absolute truths that are necessarily derived from this
reality. When considered though, this postmodern view of 'no absolute
truths' is rather strange, because people, generally, don't jump
out of windows thinking they can fly, as they recognise this absolute truth
of gravity and the resultant risk of injury or death. And there are many
examples of these absolute truths in daily life that we must abide by to
survive, e.g. sleeping, eating, breathing, etc.
So my first point is that there are absolute truths, founded
on physical reality, and these are clearly important
for our survival.
However, of profoundly more importance is that I am now quite convinced that the source of this absolute truth, i.e. physical reality itself, has finally been discovered (i.e. we have finally worked out the correct language for describing how matter exists and moves about in Space). That careful study of these subjects of Philosophy, Physics and Metaphysics over the past ten years has convinced me (and many others) that there is a very simple and sensible way of describing reality that clearly corrects a lot of past errors and problems of human knowledge.
Basically summarised, the current paradigm of representing matter as discrete
'particles' that generate 'fields' in
'space-time', while useful, is only an approximation of reality,
and it causes numerous problems because of this. (This is well known, and
explains the academic foundations of our postmodern culture of no absolute
truths).
To correct these errors it is necessary to reject the 'particle' conception
of matter (as Einstein did, see below) and describe matter in terms of Spherical
Standing Waves in Space that cause the particle effect at their
Wave-Center.
Effectively we are combining the Absolute Space assumed
by Newton (1678) with the spherically spatially
extended structure of matter as assumed by Albert Einstein
in his Theory of Relativity (1905 - 1916) and
the scalar wave properties of matter discovered by Schrodinger
and de Broglie (foundations of Quantum Theory,
1928).
Two further points are important here to correctly understand these central
concepts of the Wave Structure of Matter.
Firstly, Newton's Absolute Space was considered a 'background' reference
frame for the motion (and acceleration) of matter 'particles'. Thus in Newton's
Space matter did not affect Space (matter was somehow separate as 'particles').
Einstein rejected the 'particle' conception of matter and tried to unite
matter and Space (and time, gravity) as one thing, by representing matter
as continuous spherical fields. So in Einstein's relativity matter does
affect Space, as matter and space are united (i.e. matter is spherically
spatially extended and represented as a spherical field).
The Wave Structure of Matter agrees with Einstein, Matter and Space are
one and the same thing (there are no 'particles'), and thus matter does
affect space and its properties. The central difference is we are describing
matter in terms of Spherical Waves in Continuous Space,
rather than Einstein's (failed) field theory of matter as Continuous
Spherical Fields in Space-Time.
Secondly, and this is mainly to physicists, it is important to recognise that there are two very different types of waves used in physics, the vector electromagnetic waves developed by Maxwell, which describe both a quantity and direction of force, and the Scalar waves of Quantum Theory, which are described by a Wave Amplitude only. The Wave Structure of Matter, which describes matter as Spherical Waves in a physically real Space, requires the use of the Scalar Waves from Quantum Theory (as Physicists would know, there are no spherical solutions for vector electromagnetic waves - which is why Richard Feynman had such problems!)
The Wave Structure of Matter is explained in more detail in the short summary to physics below. And while I do realise that new knowledge is generally confusing to begin (a limitation of the human mind that affects us all), I can assure you, that once understood, the Wave Structure of Matter is surprisingly simple, and very obvious and sensible.
Significantly, we are describing reality from One thing, Space, and its properties as a Wave Medium, rather than from Many things, Matter, and their properties of generating Fields. There are two reasons why this unity of reality is important.
Firstly, Metaphysics is founded on the Principle that the many things we experience must somehow be inter-connected by One thing that causes and connects them. And this is not an abstract academic idea, it is just confirming our experience of the world (look around you and think about how you are connected to these other objects that you see in Space). I have many quotes on this unity of reality (it is the central foundation of both philosophy and metaphysics since the time of Ancient Greek and Indian philosophy) but Leibniz summarises it most concisely.
Reality cannot be found except in One single source, because of the interconnection of all things with one another. (Gottfried Leibniz, 1670)
Secondly, and this relates to Science, there is the fundamental principle of simplicity, Occam's razor, where the theory that describes the most things from the least assumptions is the best theory. And it is clear to anyone who takes the time to consider this, that the Wave Structure of Matter, by being founded on One thing existing, Space, and its wave motions that form matter, is the most simple way of describing reality. The scientific measure of its truth is to then show that it deduces correctly what we observe from observation and experiment, which then takes us to the pioneering work of mathematical physicist Dr Milo Wolff, who has deduced many of the central laws of modern physics from this most simple foundation for the Sciences, the Wave Structure of Matter.
Nonetheless, I realise that what I believe and write about reality is pretty
meaningless and irrelevant to the vast majority of people. Considering that
the internet is full of well meaning crackpots, combined with the current
complexity / diversity of knowledge, and the past failures to correctly
describe Reality, it is pretty obvious few people will take this seriously
(or make any effort to understand it to determine its truth - and there
are many valid reasons for this apparent apathy and cynicism.)
I believe though, that the best I can do as a philosopher is to carefully
explain this knowledge as simply and sensibly as possible, and perhaps more
importantly, explain why knowledge of reality and its resultant absolute
truths is critically important to our future survival (particularly now,
with the many problems that currently confront humanity).
Though I have read many wonderful minds (I admit to being fascinated by
the history and evolution of human knowledge) I think David Bohm sums up
the reasons why this knowledge of the unity of reality is fundamentally
important to humanity, as he writes;
The notion that all these fragments is separately existent
is evidently an illusion, and this illusion cannot do other than lead to
endless conflict and confusion. Indeed, the attempt to live according to
the notion that the fragments are really separate is, in essence, what has
led to the growing series of extremely urgent crises that is confronting
us today. Thus, as is now well known, this way of life has brought about
pollution, destruction of the balance of nature, over-population, world-wide
economic and political disorder and the creation of an overall environment
that is neither physically nor mentally healthy for most of the people who
live in it. Individually there has developed a widespread feeling of helplessness
and despair, in the face of what seems to be an overwhelming mass of disparate
social forces, going beyond the control and even the comprehension of the
human beings who are caught up in it.
(David Bohm, Wholeness and the Implicate Order, 1980)
The real problem that this new knowledge faces is that it contradicts
the current post-modern belief that it is impossible to know Reality, and
thus to have Absolute Truth. All knowledge is relative to cultural constructions,
which are ultimately merely ideas, not real things existing in themselves
(Kantian
Idealism). I suspect it would be 'academic suicide' to even use these
terms of Truth and Reality in a title as I have done (though they accurately
reflect the content). You are asking to be branded a 'crackpot', either
ignorant of our current 'enlightened post-modern' knowledge, and thus deluded,
or allowing ego and imagination to sway reason and good sense.
In my defense, which is really a defense of Philosophy and Science, I reply;
Over the past ten years I have taken the time to study the great minds of
human history (there are many beautiful and brilliant minds to read). This
becomes clear as you read this website, which is filled with thousands of
quotes from many great minds over thousands of years.
Now this in no way means that what I write is true, my point is more that
by presenting the existing knowledge from the original source, explaining
the problems and how they can be solved, this then allows you to make up
your own mind as to the validity of the argument. (As I see philosophy and
science, you can't tell someone the truth, but you can show them how to
work it out for themselves.)
And based upon this knowledge of philosophy, physics and metaphysics, it
is now clear to me that the Wave Structure of Matter is a far better way
to describe Reality than the current paradigm of particles and fields in
space and time.
Clearly, if this knowledge of the Wave Structure of Matter is correct,
then it is very important to Humanity, and has the power to solve many of
our problems, both in the spheres of human knowledge, and how we are to
live on Earth (without destroying ourselves and the Nature which created
us and upon which we depend). As I see it, the world needs our help, and
this help must be founded on the Truth, not just our good intentions, if
it is to actually work and make things better.
In a busy world I urge people to take the time to read and think about the
Wave Structure of Matter. Truth and Reality are not only fascinating subjects,
they describe what we really are as Humans, how we have evolved to exist
as matter in this Space of the universe, and thus offer great wisdom as
to how we should live here on our fragile and most amazing little planet,
our dear Earth and its wonderful diversity of life.
So there you have it - the end of the serious lecture / introduction (sorry!).
My hope is that it encourages you to take the time to read the following
summary of the Wave Structure of Matter (which is only a couple of pages
long and i think it is very useful!). You will also find in this website
many articles on philosophy, physics and metaphysics that I believe explain
and solve most of the major / central problems of these fields of knowledge.
And yes, I appreciate that this claim clearly falls in the class of 'crackpot',
however it is also the obvious consequence of correctly knowing physical
reality. So i hope you will think about it!
Sincerely,
Geoff Haselhurst
Email,
(December 2005).
Short Summary of Physics and the Wave Structure of Matter in Space
At a fundamental level Modern Physics is founded on the concepts of particles and fields in space and time. Further difficulties arise because both light and matter exhibit a particle / wave duality, and there are two main fields, charge (electromagnetic) and mass (gravitational / inertial). While this is still a little simplistic, it makes the point that we have a number of different concepts without any clear understanding of how they are connected. This causes numerous problems for Human knowledge (and society), as we do not understand the necessary connections between these many things we experience. And as Hume made clear (see David Hume's Problem of Causation and Necessary Connection), without this knowledge of necessary connection between things that exist we can have no causal theory of knowledge, leaving the Sciences founded on Induction from repeated observation which is always uncertain (rather than deduction from metaphysical principles which is certain);
Experience only teaches us, how one event constantly follows another; without instructing us in the secret connexion, which binds them together, and renders them inseparable. (David Hume)
So how do we solve these problems? I think the simplest way to explain this is to list these central concepts of physics, show which path Albert Einstein took to try and unite them from a common foundation (which he failed to do), and then demonstrate from the most simple foundation (founded on One thing Space existing as a Wave Medium) how we can finally unite these concepts in a meaningful way that describes reality without paradox or contradiction. Now I know that most of you will be very skeptical of such a big claim, but the solution is pretty simple and obvious once known so I hope you will persevere (and it is concise / short!).
Central Concepts of Modern Physics
Newton's Mechanics - Space,
Time, Matter as Particles
with 'Mass', Force = Mass
by Acceleration.
Continuous Electromagnetic Field Theory (Faraday,
Maxwell, Lorentz, Einstein / Relativity) - Matter as Particles with 'Charge',
Continuous Spherical Electromagnetic Fields,
Light as Vector Electromagnetic Waves, Gravitational
Fields (local theory - all matter interactions limited by velocity
of light c)
Discrete Quantum Theory (Planck, Einstein, de Broglie,
Schrodinger, Born) - Light as Discrete Particles
(Photons), Scalar Waves,
Probability Waves, Particle
Wave Duality (Light & Matter), Quantum Entanglement
(apparently non local theory - appear to be instantaneous matter interactions).
While this is still a little simplistic, you can see how confusing Physics becomes when you have so many concepts, some of which clearly contradict others (e.g. light and matter behave as both particles and waves, discrete particles and continuous fields, local and non local interactions). Albert Einstein attempted to simplify this mess with his 'field theory of matter' where he discarded the 'particle' concept and tried to represent matter as spherical fields in space-time, as he writes;
Physical objects are not in space, but these objects are spatially extended (as fields). In this way the concept 'empty space' loses its meaning. ... The field thus becomes an irreducible element of physical description, irreducible in the same sense as the concept of matter (particles) in the theory of Newton. ... The physical reality of space is represented by a field whose components are continuous functions of four independent variables - the co-ordinates of space and time. Since the theory of general relativity implies the representation of physical reality by a continuous field, the concept of particles or material points cannot play a fundamental part, nor can the concept of motion. The particle can only appear as a limited region in space in which the field strength or the energy density are particularly high. (Albert Einstein, 1950)
When forced to summarize the general theory
of relativity in one sentence:
Time and space and gravitation
have no separate existence from matter. (Albert Einstein)
Now there are a number of Problems with Albert Einstein's Field Theory of Matter, as he was well aware, and which, late in his life, caused him to write to his friend Michael Besso expressing his frustration;
All these fifty years of conscious brooding have brought me no nearer to the answer to the question, 'What are light quanta?' Nowadays every Tom, Dick and Harry thinks he knows it, but he is mistaken. … I consider it quite possible that physics cannot be based on the field concept, i.e., on continuous structures. In that case, nothing remains of my entire castle in the air, gravitation theory included, [and of] the rest of modern physics. (Albert Einstein, 1954)
In hindsight it is easier to understand Einstein's error - the clue is that Quantum Theory's discovery of the wave properties of matter did not occur until 1928, whereas Einstein's continuous electromagnetic field foundations were developed from 1905 to 1916 based largely on the ideas of James Clerk Maxwell (Maxwell's Equations, 1870s) and Lorentz's Theory of the Electron (1900). So basically his mistake was to work with fields in space-time (mathematical) rather than real waves in Space (physical), largely because he did not have knowledge of the Wave properties of matter when he developed his Theory of Relativity.
In terms of the Metaphysical
foundations of Physics, the central error has been to try and describe
reality from the Many material things we experience, matter
'particles'. (i.e. Science, which is empirically founded). And even though
Einstein rejected the 'particle', his field theory of matter is still founded
on this priority of matter (that matter's field interactions cause the effect
of space and time).
However, Metaphysics has known for several thousand years that it is necessary
to describe reality from One thing existing to explain
matter's interconnection in Space and the Universe. As Aristotle, Leibniz
and Bradley write;
The first philosophy (Metaphysics) is universal
and is exclusively concerned with primary substance. ... And here we will
have the science to study that which is, both in its essence and in the
properties which, just as a thing that is, it has. (Aristotle,
340BC)
Reality cannot be found except in One single source, because of the interconnection
of all things with one another. ... I do not conceive of any reality at
all as without genuine unity. (Gottfried Leibniz, 1670)
We may agree, perhaps, to understand by Metaphysics an attempt to know reality
as against mere appearance, or the study of first principles or ultimate
truths, or again the effort to comprehend the universe, not simply piecemeal
or by fragments, but somehow as a whole. (Bradley, 1846-1924)
By abiding by this central rule of Metaphysics and describing Reality in Terms of One thing existing, Space, and its Properties as a Wave Medium for Spherical Standing Waves that form Matter, we find a very simple solution to all of the above problems of Physics (again this is a big claim, but this website does explain many of these things). Basically Einstein is correct, Matter is a Spherically Spatially extended structure of Space (there is no 'particle') though most importantly we have simplified Einstein's ideas from;
Matter as Continuous Spherical Fields in Space-Time
to
Matter as Spherical Waves in Continuous Space.
![]() +
+ ![]() =
= ![]() This rough diagram shows how the In-Waves and Out-Waves form a Standing
Wave around the Wave-Center 'particle'.
This rough diagram shows how the In-Waves and Out-Waves form a Standing
Wave around the Wave-Center 'particle'.
Significantly, we have now decreased the number of concepts from Einstein's 5 (continuous, spherical, fields, space, time) where there is no clear understanding of how they are connected, down to 4 concepts (spherical, waves, continuous, space) where we can clearly connect these four things back to One thing existing, Space, and its Properties as a Wave Medium for Spherical Waves that form matter. And after you have studied philosophy and metaphysics for a while, you come to realise that this solution to the most central and profound of all problems, the Problem of the One and the Many, of how the many things are caused and connected by the One thing then enables you to explain and solve numerous other problems that this lack of knowledge of necessary connection caused.
And from this dynamic unity of reality it is quite simple to show that:
The discrete 'particle' effect of matter
is caused by the Wave-Center of the Spherical Standing
Waves (the diagram shows that this is an obvious solution to the particle
/ wave duality of matter!).
The discrete 'particle' effect of light
is caused by discrete Standing Wave Interactions / Resonant
Coupling.
Time is caused
by Wave Motion (as spherical wave motions of Space which
cause matter's activity and the phenomena of time).
Forces / Fields are caused by wave interactions of the
Spherical In and Out Waves with other matter in the universe which change
the location of the Wave-Center (and which we 'see' as a 'force accelerating
a particle').
Quantum Entanglement is likewise caused by the Interaction
between the In and Out-Waves and all the other matter in the universe, thus
matter is always subtly connected to other matter in the universe (i.e.
matter is large not small, we only see the Wave-Center and have been deceived
by its 'particle' effect). However, Einstein's 'Locality' is correct, all
matter to matter interactions are limited by the velocity of Waves in Space.
Of most significance (the real beauty of this solution) is that not only does it solve the problems of Albert Einstein's Theory of Relativity, but it also solves the basic problems of Quantum Theory / Quantum Mechanics (founded on Wave Equations) and Cosmology (uniting finite matter & universe with infinite eternal space - The Big Bang Theory being founded on a basic error).
Geoff Haselhurst (January, 2005)
Change the World with Truth and Reality
Below are some relevant quotes that we hope you find interesting / illuminating! The three central points are;
i) The underlying dynamic unity of reality (which the Wave Structure of Matter in Space clearly explains) has been known for thousands of years (though this has largely been ignored by western scientists, Einstein, Schrodinger and Bohm are the most famous exceptions).
ii) If you want to change / improve the world, well it is up to you, and it requires knowledge of truth, and thus reality (good intentions are not good enough - in fact they just lead to hell on earth).
iii) The discovery of reality is obviously the most profound knowledge
that we can have, and given human nature, it is also the most difficult
to get fairly considered by science / society. Few people have enough knowledge
to judge it sensibly / logically, and those who do tend to be corrupted
by the current postmodern view of 'no absolute truths' thus ignore it. (Which
is why we need your help in getting these pages to rank well on the Internet!)
 (Rachel
Carson) We stand now where two roads diverge. But unlike the
roads in Robert Frost's familiar poem, they are not equally fair. The
road we have long been traveling is deceptively easy, a smooth superhighway
on which we progress with great speed, but at its end lies disaster.
The other fork of the road - the one 'less traveled by' - offers our
last, our only chance to reach a destination that assures the preservation
of the earth. The choice, after all, is ours to make. (Rachel
Carson) We stand now where two roads diverge. But unlike the
roads in Robert Frost's familiar poem, they are not equally fair. The
road we have long been traveling is deceptively easy, a smooth superhighway
on which we progress with great speed, but at its end lies disaster.
The other fork of the road - the one 'less traveled by' - offers our
last, our only chance to reach a destination that assures the preservation
of the earth. The choice, after all, is ours to make. |
 (Margaret
Mead) Never doubt that a small group of thoughtful committed
citizens can change the world, indeed it is the only thing that ever
has. (Margaret
Mead) Never doubt that a small group of thoughtful committed
citizens can change the world, indeed it is the only thing that ever
has. |
 (Albert
Einstein) The free, unhampered exchange of ideas and scientific
conclusions is necessary for the sound development of science, as it
is in all spheres of cultural life. ... (Albert
Einstein) The free, unhampered exchange of ideas and scientific
conclusions is necessary for the sound development of science, as it
is in all spheres of cultural life. ...We must not conceal from ourselves that no improvement in the present depressing situation is possible without a severe struggle; for the handful of those who are really determined to do something is minute in comparison with the mass of the lukewarm and the misguided. ... Humanity is going to need a substantially new way of thinking if it is to survive. When forced to summarize the general theory of relativity in one sentence: Time and space and gravitation have no separate existence from matter. ... Physical objects are not in space, but these objects are spatially extended (as fields). In this way the concept 'empty space' loses its meaning. ... The field thus becomes an irreducible element of physical description, irreducible in the same sense as the concept of matter (particles) in the theory of Newton. ... The physical reality of space is represented by a field whose components are continuous functions of four independent variables - the co-ordinates of space and time. Since the theory of general relativity implies the representation of physical reality by a continuous field, the concept of particles or material points cannot play a fundamental part, nor can the concept of motion. The particle can only appear as a limited region in space in which the field strength or the energy density are particularly high. The subtlety of the concept of space was enhanced by the discovery that there exist no completely rigid bodies. All bodies are elastically deformable. |
 (Erwin
Schrodinger) What we observe as material bodies and forces
are nothing but shapes and variations in the structure of space. Particles
are just schaumkommen (appearances). ... The world is given to me only
once, not one existing and one perceived. Subject and object are only
one. The barrier between them cannot be said to have broken down as
a result of recent experience in the physical sciences, for this barrier
does not exist. ... Let me say at the outset, that in this discourse,
I am opposing not a few special statements of quantum mechanics held
today (1950s), I am opposing as it were the whole of it, I am opposing
its basic views that have been shaped 25 years ago, when Max Born put
forward his probability interpretation, which was accepted by almost
everybody. ... I don't like it, and I'm sorry I ever had anything to
do with it. ... The scientist only imposes two things, namely truth
and sincerity, imposes them upon himself and upon other scientists. (Erwin
Schrodinger) What we observe as material bodies and forces
are nothing but shapes and variations in the structure of space. Particles
are just schaumkommen (appearances). ... The world is given to me only
once, not one existing and one perceived. Subject and object are only
one. The barrier between them cannot be said to have broken down as
a result of recent experience in the physical sciences, for this barrier
does not exist. ... Let me say at the outset, that in this discourse,
I am opposing not a few special statements of quantum mechanics held
today (1950s), I am opposing as it were the whole of it, I am opposing
its basic views that have been shaped 25 years ago, when Max Born put
forward his probability interpretation, which was accepted by almost
everybody. ... I don't like it, and I'm sorry I ever had anything to
do with it. ... The scientist only imposes two things, namely truth
and sincerity, imposes them upon himself and upon other scientists. |
 (David
Bohm, Wholeness and the Implicate Order, 1980) The notion that
all these fragments is separately existent is evidently an illusion,
and this illusion cannot do other than lead to endless conflict and
confusion. Indeed, the attempt to live according to the notion that
the fragments are really separate is, in essence, what has led to the
growing series of extremely urgent crises that is confronting us today.
Thus, as is now well known, this way of life has brought about pollution,
destruction of the balance of nature, over-population, world-wide economic
and political disorder and the creation of an overall environment that
is neither physically nor mentally healthy for most of the people who
live in it. Individually there has developed a widespread feeling of
helplessness and despair, in the face of what seems to be an overwhelming
mass of disparate social forces, going beyond the control and even the
comprehension of the human beings who are caught up in it. (David
Bohm, Wholeness and the Implicate Order, 1980) The notion that
all these fragments is separately existent is evidently an illusion,
and this illusion cannot do other than lead to endless conflict and
confusion. Indeed, the attempt to live according to the notion that
the fragments are really separate is, in essence, what has led to the
growing series of extremely urgent crises that is confronting us today.
Thus, as is now well known, this way of life has brought about pollution,
destruction of the balance of nature, over-population, world-wide economic
and political disorder and the creation of an overall environment that
is neither physically nor mentally healthy for most of the people who
live in it. Individually there has developed a widespread feeling of
helplessness and despair, in the face of what seems to be an overwhelming
mass of disparate social forces, going beyond the control and even the
comprehension of the human beings who are caught up in it. |
 (Lee
Smolin, 1997) A successful unification of quantum theory and
relativity would necessarily be a theory of the universe as a whole.
It would tell us, as Aristotle and Newton did before, what space and
time are, what the cosmos is, what things are made of, and what kind
of laws those things obey. Such a theory will bring about a radical
shift - a revolution - in our understanding of what nature is. It must
also have wide repercussions, and will likely bring about, or contribute
to, a shift in our understanding of ourselves and our relationship to
the rest of the universe. (Lee
Smolin, 1997) A successful unification of quantum theory and
relativity would necessarily be a theory of the universe as a whole.
It would tell us, as Aristotle and Newton did before, what space and
time are, what the cosmos is, what things are made of, and what kind
of laws those things obey. Such a theory will bring about a radical
shift - a revolution - in our understanding of what nature is. It must
also have wide repercussions, and will likely bring about, or contribute
to, a shift in our understanding of ourselves and our relationship to
the rest of the universe. |
|
 (Charles
Darwin) Although I am fully convinced of the truth of the views
given in this volume I by no means expect to convince experienced naturalists
whose minds are stocked with a multitude of facts all viewed, during
a long course of years, from a point of view directly opposite to mine.
But I look with confidence to the future to young and rising naturalists,
who will be able to view both sides of the question with impartiality. (Charles
Darwin) Although I am fully convinced of the truth of the views
given in this volume I by no means expect to convince experienced naturalists
whose minds are stocked with a multitude of facts all viewed, during
a long course of years, from a point of view directly opposite to mine.
But I look with confidence to the future to young and rising naturalists,
who will be able to view both sides of the question with impartiality. |
 (Galileo
Galilei, 1600) I wish, my dear Kepler, that we could have a
good laugh together at the extraordinary stupidity of the mob. What
do you think of the foremost philosophers of this University? In spite
of my oft-repeated efforts and invitations, they have refused, with
the obstinacy of a glutted adder, to look at the planets or Moon or
my telescope. ... (Galileo
Galilei, 1600) I wish, my dear Kepler, that we could have a
good laugh together at the extraordinary stupidity of the mob. What
do you think of the foremost philosophers of this University? In spite
of my oft-repeated efforts and invitations, they have refused, with
the obstinacy of a glutted adder, to look at the planets or Moon or
my telescope. ...In questions of science, the authority of a thousand is not worth the humble reasoning of a single individual. |
 (Karl
Popper, 1975) In my opinion, the greatest scandal of philosophy
is that, while all around us the world of nature perishes - and not
the world of nature alone - philosophers continue to talk, sometimes
cleverly and sometimes not, about the question of whether this world
exists. They get involved in scholasticism, in linguistic puzzles such
as, for example, whether or not there are differences between 'being'
and 'existing'. ... Denying realism amounts to megalomania (the most
widespread occupational disease of the professional philosopher). ...
If a theory corresponds to the facts but does not cohere with some earlier
knowledge, then this earlier knowledge should be discarded. (Karl
Popper, 1975) In my opinion, the greatest scandal of philosophy
is that, while all around us the world of nature perishes - and not
the world of nature alone - philosophers continue to talk, sometimes
cleverly and sometimes not, about the question of whether this world
exists. They get involved in scholasticism, in linguistic puzzles such
as, for example, whether or not there are differences between 'being'
and 'existing'. ... Denying realism amounts to megalomania (the most
widespread occupational disease of the professional philosopher). ...
If a theory corresponds to the facts but does not cohere with some earlier
knowledge, then this earlier knowledge should be discarded. |
 (Bertrand
Russell) Thought is subversive and revolutionary, destructive and terrible,
Thought is merciless to privilege, established institutions, and comfortable
habit. Thought is great and swift and free. ... (Bertrand
Russell) Thought is subversive and revolutionary, destructive and terrible,
Thought is merciless to privilege, established institutions, and comfortable
habit. Thought is great and swift and free. ...The whole problem with the world is that fools and fanatics are always so certain of themselves, and wiser people so full of doubts. ... Do not fear to be eccentric in opinion, for every opinion now accepted was once eccentric. ... Many people would sooner die than think; in fact, they do so. ... It has been said that man is a rational animal. All my life I have been searching for evidence which could support this. |
 (Friedrich
Nietzsche, The Greeks) Greek philosophy seems to begin with
a preposterous fancy, with the proposition that water is the origin
and mother-womb of all things. Is it really necessary to stop there
and become serious? Yes, and for three reasons: firstly, because the
preposition does enunciate something about the origin of things; secondly,
because it does so without figure and fable; thirdly and lastly, because
it contained, although only in the chrysalis state, the idea :everything
is one. ... That which drove him (Thales) to this generalization was
a metaphysical dogma, which had its origin in a mystic intuition and
which together with the ever renewed endeavors to express it better,
we find in all philosophies- the proposition: everything is
one! ... (Friedrich
Nietzsche, The Greeks) Greek philosophy seems to begin with
a preposterous fancy, with the proposition that water is the origin
and mother-womb of all things. Is it really necessary to stop there
and become serious? Yes, and for three reasons: firstly, because the
preposition does enunciate something about the origin of things; secondly,
because it does so without figure and fable; thirdly and lastly, because
it contained, although only in the chrysalis state, the idea :everything
is one. ... That which drove him (Thales) to this generalization was
a metaphysical dogma, which had its origin in a mystic intuition and
which together with the ever renewed endeavors to express it better,
we find in all philosophies- the proposition: everything is
one! ...There is nothing more necessary than truth, and in comparison with it everything else has only secondary value. This absolute will to truth: what is it? Is it the will to not allow ourselves to be deceived? Is it the will not to deceive? One does not want to be deceived, under the supposition that it is injurious, dangerous, or fatal to be deceived. ... Do not allow yourselves to be deceived: Great Minds are Skeptical. What if God were not exactly truth, and if this could be proved? And if he were instead the vanity, the desire for power, the ambitions, the fear, and the enraptured and terrified folly of mankind? (Nietzsche, 1890) |
 (Arthur
Schopenhauer) It is a ticklish question, the steering of the
public, good and docile as it is on the whole. Although as a rule the
absurd culminates, and it seems impossible for the voice of the individual
ever to penetrate through the chorus of foolers and fooled, still there
is left to the genuine works of all times a quite peculiar, silent,
slow, and powerful influence; and as if by a miracle, we see them rise
at last out of the turmoil like a balloon that floats up out of the
thick atmosphere of this globe into purer regions. Having once arrived
there, it remains at rest, and no one can any longer draw it down again.
... (Arthur
Schopenhauer) It is a ticklish question, the steering of the
public, good and docile as it is on the whole. Although as a rule the
absurd culminates, and it seems impossible for the voice of the individual
ever to penetrate through the chorus of foolers and fooled, still there
is left to the genuine works of all times a quite peculiar, silent,
slow, and powerful influence; and as if by a miracle, we see them rise
at last out of the turmoil like a balloon that floats up out of the
thick atmosphere of this globe into purer regions. Having once arrived
there, it remains at rest, and no one can any longer draw it down again.
...The task is not so much to see what no one yet has seen, but to think what no body yet has thought about that which everyone sees. ... For the incredibly great majority of men are by their nature absolutely incapable of any but material aims; they cannot even comprehend any others. Accordingly, the pursuit of truth alone is a pursuit far too lofty and eccentric for us to expect that all or many, or indeed even a mere few, will sincerely take part in it. ... But life is short, and truth works far and lives long: let us speak the truth. (Arthur Schopenhauer, 1818) |
 (Immanuel
Kant, 1781) It is the duty of philosophy to destroy the illusions
which had their origin in misconceptions, whatever darling hopes and
valued expectations may be ruined by its explanations. ... and thus
to prevent the scandal which metaphysical controversies are sure, sooner
or later, to cause even to the masses. (Immanuel
Kant, 1781) It is the duty of philosophy to destroy the illusions
which had their origin in misconceptions, whatever darling hopes and
valued expectations may be ruined by its explanations. ... and thus
to prevent the scandal which metaphysical controversies are sure, sooner
or later, to cause even to the masses. |
 (David Hume, 1737) And though the philosopher may live
remote from business, the genius of philosophy, if carefully cultivated
by several, must gradually diffuse itself throughout the whole society,
and bestow a similar correctness on every art and calling.
(David Hume, 1737) And though the philosopher may live
remote from business, the genius of philosophy, if carefully cultivated
by several, must gradually diffuse itself throughout the whole society,
and bestow a similar correctness on every art and calling.When we look about us towards external objects, and consider the operation of causes, we are never able, in a single instance, to discover any power or necessary connexion; any quality, which binds the effect to the cause, and renders the one an infallible consequence of the other. There is required a medium, which may enable the mind to draw such an inference, if indeed it be drawn by reasoning and argument. What that medium is, I must confess, passes my comprehension; and it is incumbent on those to produce it, who assert that it really exists, and is the origin of all our conclusions concerning matter of fact. This question I propose as much for the sake of information, as with an intention of raising difficulties. I cannot find, I cannot imagine any such reasoning. But I keep my mind still open to instruction, if any one will vouchsafe to bestow it upon me. We find in the course of nature that though the effects be many, the principles from which they arise are commonly few and simple, and that it is the sign of an unskilled naturalist to have recourse to a different quality in order to explain every different operation. |
 (Gottfried
Leibniz, 1670) I agree with you that it is important to examine
our presuppositions, thoroughly and once for all, in order to establish
something solid. For I hold that it is only when we can prove all that
we bring forward that we perfectly understand the thing under consideration.
I know that the common herd takes little pleasure in these researches,
but I know also that the common herd take little pains thoroughly to
understand things. ... a distinction must be made between true and false
ideas, and that too much rein must not be given to a man's imagination
under pretext of its being a clear and distinct intellection. ... Reality
cannot be found except in One single source, because of the interconnection
of all things with one another. ... I do not conceive of any reality
at all as without genuine unity. ... I maintain also that substances,
whether material or immaterial, cannot be conceived in their bare essence
without any activity, activity being of the essence of substance
in general. (Gottfried
Leibniz, 1670) I agree with you that it is important to examine
our presuppositions, thoroughly and once for all, in order to establish
something solid. For I hold that it is only when we can prove all that
we bring forward that we perfectly understand the thing under consideration.
I know that the common herd takes little pleasure in these researches,
but I know also that the common herd take little pains thoroughly to
understand things. ... a distinction must be made between true and false
ideas, and that too much rein must not be given to a man's imagination
under pretext of its being a clear and distinct intellection. ... Reality
cannot be found except in One single source, because of the interconnection
of all things with one another. ... I do not conceive of any reality
at all as without genuine unity. ... I maintain also that substances,
whether material or immaterial, cannot be conceived in their bare essence
without any activity, activity being of the essence of substance
in general. |
 (George
Berkeley) Nothing seems of more importance, towards erecting
a firm system of sound and real knowledge, which may be proof against
the assaults of scepticism, than to lay the beginning in a distinct
explication of what is meant by thing, reality, existence: for in vain
shall we dispute concerning the real existence of things, or pretend
to any knowledge thereof, so long as we have not fixed the meaning of
those words. ... we are under an invincible blindness as to the true
and real nature of things. ... (George
Berkeley) Nothing seems of more importance, towards erecting
a firm system of sound and real knowledge, which may be proof against
the assaults of scepticism, than to lay the beginning in a distinct
explication of what is meant by thing, reality, existence: for in vain
shall we dispute concerning the real existence of things, or pretend
to any knowledge thereof, so long as we have not fixed the meaning of
those words. ... we are under an invincible blindness as to the true
and real nature of things. ...Hence a great number of dark and ambiguous terms presumed to stand for abstract notions, have been introduced into metaphysics and morality, and from these have grown infinite distractions and disputes amongst the learned. ... My purpose therefore is, to try if I can discover what those principles are, which have introduced all that doubtfulness and uncertainty, those absurdities and contradictions into the several sects of philosophy; insomuch that the wisest men have thought our ignorance incurable, conceiving it to arise from the natural dullness and limitation of our faculties. ... |
 (Spinoza)
I have striven not to laugh at human actions, not to weep at them, nor
to hate them, but to understand them. ... (Spinoza)
I have striven not to laugh at human actions, not to weep at them, nor
to hate them, but to understand them. ... But if men would give heed to the nature of substance they would doubt less concerning the Proposition that Existence appertains to the nature of substance: rather they would reckon it an axiom above all others, and hold it among common opinions. ... A substance cannot be produced from anything else: it will therefore be its own cause, that is, its essence necessarily involves existence, or existence appertains to the nature of it. ... No two or more substances can have the same attribute and it appertains to the nature of substance that it should exist. It must therefore exist finitely or infinitely. But not finitely. For it would then be limited by some other substance of the same nature which also of necessity must exist: and then two substances would be granted having the same attribute, which is absurd. It will exist, therefore, infinitely. |
 (Aristotle)
The first philosophy (Metaphysics) is universal and is exclusively concerned
with primary substance. ... And here we will have the science to study
that which is just as that which is, both in its essence and in the
properties which, just as a thing that is, it has. (Aristotle)
The first philosophy (Metaphysics) is universal and is exclusively concerned
with primary substance. ... And here we will have the science to study
that which is just as that which is, both in its essence and in the
properties which, just as a thing that is, it has. The entire preoccupation of the physicist is with things that contain within themselves a principle of movement and rest. And to seek for this is to seek for the second kind of principle, that from which comes the beginning of the change. There must then be a principle of such a kind that its substance is activity. ... it is impossible that the primary existent, being eternal, should be destroyed. ... that among entities there must be some cause which moves and combines things. ..about its coming into being and its doings and about all its alterations we think that we have knowledge when we know the source of its movement. (Aristotle, Metaphysics) |
 (Plato,
Republic) And those whose hearts are fixed on Reality
itself deserve the title of Philosophers. ... (Plato,
Republic) And those whose hearts are fixed on Reality
itself deserve the title of Philosophers. ... The society we have described can never grow into a reality or see the light of day, and there will be no end to the troubles of states, or indeed, my dear Glaucon, of humanity itself, till philosophers are kings in this world, or till those we now call kings and rulers really and truly become philosophers, and political power and philosophy thus come into the same hands, while the many natures now content to follow either to the exclusion of the other are forcibly debarred from doing so. This is what I have hesitated to say so long, knowing what a paradox it would sound; for it is not easy to see that there is no other road to happiness, either for society or the individual. ... When the mind's eye rests on objects illuminated by truth and reality, it understands and comprehends them, and functions intelligently; but when it turns to the twilight world of change and decay, it can only form opinions, its vision is confused and its beliefs shifting, and it seems to lack intelligence. (Plato, Republic) |
 (Socrates)
Know Thyself, the unexamined life is not worth living. ... Wisdom begins
in wonder. ... There is only one good, knowledge, and one evil, ignorance.
... Beware the barrenness of a busy life. ... (Socrates)
Know Thyself, the unexamined life is not worth living. ... Wisdom begins
in wonder. ... There is only one good, knowledge, and one evil, ignorance.
... Beware the barrenness of a busy life. ... Are you not ashamed that you give your attention to acquiring as much money as possible, and similarly with honour and reputation, and care so little about wisdom and truth and the greatest improvement of the soul, which you never regard or heed at all? Wars, factions, and fighting, said Socrates as he looked forward from his last hour, have no other origin than this same body and its lusts. ... We must set the soul free from it; we must behold things as they are. And having thus got rid of the foolishness of the body, we shall be pure and hold converse with the pure, and shall in our own selves have complete knowledge of the Incorruptible which is, I take it, no other than the very truth. (Socrates, 469 - 399 B.C.) |
 (Max
Planck) A new scientific truth does not triumph by convincing
its opponents and making them see the light, but rather because its
opponents eventually die, and a new generation grows up that is familiar
with it.
(Max
Planck) A new scientific truth does not triumph by convincing
its opponents and making them see the light, but rather because its
opponents eventually die, and a new generation grows up that is familiar
with it.Help Humanity
"You must be the change you wish to see in the world."
(Mohandas Gandhi)
 "When forced to summarize the general theory of relativity in one sentence:
Time and space and gravitation have no separate existence from matter. ... Physical objects are not in space, but these objects are spatially extended. In this way the concept 'empty space' loses its meaning. ... The particle can only appear as a limited region in space in which
the field strength or the energy density are particularly high. ...
"When forced to summarize the general theory of relativity in one sentence:
Time and space and gravitation have no separate existence from matter. ... Physical objects are not in space, but these objects are spatially extended. In this way the concept 'empty space' loses its meaning. ... The particle can only appear as a limited region in space in which
the field strength or the energy density are particularly high. ...
The free, unhampered exchange of ideas and scientific conclusions is necessary for the sound development of science, as it is in all spheres
of cultural life. ... We must not conceal from ourselves that no improvement in the present depressing situation is possible without
a severe struggle; for the handful of those who are really determined to do something is minute in comparison with the mass of the lukewarm
and the misguided. ...
Humanity is going to need a substantially new way of thinking if it is to survive!" (Albert Einstein)
 Our world is in great trouble due to human behaviour founded on myths and customs that are causing the destruction of Nature and climate change. We can now deduce the most simple science theory of reality - the wave structure of matter in space. By understanding how we and everything around us are interconnected
in Space we can then deduce solutions to the fundamental problems of human knowledge in physics, philosophy, metaphysics, theology, education, health, evolution and ecology, politics and society.
Our world is in great trouble due to human behaviour founded on myths and customs that are causing the destruction of Nature and climate change. We can now deduce the most simple science theory of reality - the wave structure of matter in space. By understanding how we and everything around us are interconnected
in Space we can then deduce solutions to the fundamental problems of human knowledge in physics, philosophy, metaphysics, theology, education, health, evolution and ecology, politics and society.
This is the profound new way of thinking that Einstein
realised, that we exist as spatially extended structures of the universe - the discrete and separate body an illusion. This simply confirms the
intuitions of the ancient philosophers and mystics.
Given the current censorship in physics / philosophy of science journals (based on the standard model of particle physics / big bang cosmology) the internet is the best hope for getting new knowledge
known to the world. But that depends on you, the people who care about science and society, realise the importance of truth and reality.
It is Easy to Help!
Just click on the Social Network links below, or copy a nice image or quote you like and share it. We have a wonderful collection of knowledge from the greatest minds in human history, so people will appreciate your contributions. In doing this you will help a new generation of scientists see that there is a simple sensible explanation of physical reality - the source of truth and wisdom, the only cure for the madness of man! Thanks! Geoff Haselhurst (Updated September, 2018)
A new scientific truth does not triumph by convincing its opponents and making them see the light, but rather because its opponents eventually die, and a new generation grows up that is familiar with it. (Max Planck, 1920)
| Tweet Follow @philosophytruth | |
| Geoff | |
Connect with Geoff Haselhurst at Facebook
"All that is necessary for evil to succeed is for good people to do nothing."
(Edmund Burke)
"In a time of universal deceit - telling the truth is a revolutionary act."
(George Orwell)
"Hell is Truth Seen Too Late."
(Thomas Hobbes)












Legal Disclaimer and Privacy Policy
We support 'Fair Use' of these pages for Academic & Non Commercial use.
You are welcome to use images and text, but please reference them with a link to relevant web page on this site. Thanks!
